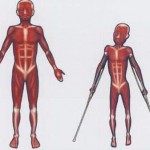
 Bệnh loạn dưỡng cơ Duchenne (DMD) là bệnh di truyền của một loại bệnh teo cơ xảy ra chủ yếu ở các bé trai. Bệnh được gây ra bởi sự đột biến trong một gen, gọi là gen DMD, có thể được di truyền lặn liên kết với X trong một gia đình, nhưng bệnh thường xảy ra ở những người kể cả khi gia đình không có tiền sử về tình trạng bệnh này. Người đã mang DMD mất dần chức năng cơ bắp và ốm yếu bắt đầu ở các chi dưới. Gen DMD là gen quy định protein dystrophin – một protein tham gia vào quá trình đảm bảo tính toàn vẹn của sợi cơ. Những bé trai với bệnh teo cơ Duchenne không sản sinh protein dystrophin trong cơ bắp của chúng.
Bệnh loạn dưỡng cơ Duchenne (DMD) là bệnh di truyền của một loại bệnh teo cơ xảy ra chủ yếu ở các bé trai. Bệnh được gây ra bởi sự đột biến trong một gen, gọi là gen DMD, có thể được di truyền lặn liên kết với X trong một gia đình, nhưng bệnh thường xảy ra ở những người kể cả khi gia đình không có tiền sử về tình trạng bệnh này. Người đã mang DMD mất dần chức năng cơ bắp và ốm yếu bắt đầu ở các chi dưới. Gen DMD là gen quy định protein dystrophin – một protein tham gia vào quá trình đảm bảo tính toàn vẹn của sợi cơ. Những bé trai với bệnh teo cơ Duchenne không sản sinh protein dystrophin trong cơ bắp của chúng.
Bệnh loạn dưỡng cơ Duchenne chiếm tỉ lệ khoảng 1 trong 3500 ca sinh bé trai trên toàn thế giới. Bởi vì đây là một rối loạn di truyền , rủi ro một gia đình bao gồm những gen của bệnh loạn dưỡng cơ Duchenne.
Các triệu chứng của bệnh loạn cơ Duchenne là gì?
Các triệu chứng thường xuất hiện trước 6 tuổi và có thể xuất hiện sớm nhất ở giai đoạn sơ sinh. Thông thường, các triệu chứng đáng chú ý đầu tiên là sự chậm phát triển của cơ vận động, bao gồm cả ngồi và đứng. Tuổi trung bình có thể đi lại ở các bé trai với bệnh loạn dưỡng cơ Duchenne là 18 tháng. Cơ chân và cơ xương chậu yếu dần, được liên kết với một khối lượng cơ bắp. Yếu cơ này gây ra một dáng đi không vững, khó leo cầu thang . Yếu cơ cũng xảy ra ở cánh tay, cổ và các khu vực khác nhưng không nghiêm trọng hoặc xuất hiện sớm ở nửa dưới của cơ thể .
Cơ bắp chân ban đầu mở rộng và các mô cơ mở rộng cuối cùng được thay thế bằng các mô mỡ và mô liên kết ( pseudohypertrophy ). Co rút cơ xảy ra ở chân, làm cho các cơ bắp không sử dụng được vì các sợi cơ rút ngắn và xơ hóa xảy ra trong mô liên kết. Đôi khi có thể gây đau ở bắp chân.
Các triệu chứng thường xuất hiện ở các bé trai tuổi từ 1-6 . Có một sự suy giảm trong sức mạnh cơ bắp trong độ tuổi từ 6 đến 11. 10 tuổi, các bé trai phải chóng nạng khi đi và khi được 12 tuổi , hầu hết các bé trai phải ngồi xe lăn. Xương phát triển bất thường, gây ra dị tật xương của cột sống và các khu vực khác.
Yếu cơ và biến dạng xương thường xuyên gây nên các rối loạn hô hấp. Bệnh cơ tim (tim to) xảy ra ở hầu hết các trường hợp, một số trường hợp bắt đầu từ tuổi thiếu niên và ở tất cả các trường hợp sau 18 tuổi. Suy giảm trí tuệ có thể xảy ra, nhưng có thể tránh khỏi và không trầm trọng như rối loạn phát triển cơ.
Vài cá nhân với DMD sống qua tuổi 30. Các biến chứng hô hấp và bệnh cơ tim là nguyên nhân phổ biến dẫn đến tử vong.
Làm thế nào để chẩn đoán bệnh loạn dưỡng cơ Duchenne?
Bệnh di truyền loạn dưỡng cơ Duchenne được chẩn đoán theo nhiều cách. Chẩn đoán lâm sàng có thể được thực hiện khi một cậu bé có tiến triển yếu cơ. Các triệu chứng xuất hiện trước 5 tuổi và chúng thường có mức độ máu creatine kinase rất cao (được mô tả dưới đây). Nếu không được điều trị, các bé trai bị bệnh phải ngồi xe lăn trước tuổi năm 13 tuổi.
Sinh thiết cơ (lấy một mẫu của cơ bắp) cho các nghiên cứu dystrophin có thể được thực hiện để tìm kiếm mức độ bất thường của dystrophin trong cơ. Protein dystrophin có thể được hình dung bằng cách nhuộm mẫu cơ bắp với một thuốc nhuộm đặc biệt cho phép ta xem các protein dystrophin. Một cơ trong đó có số lượng trung bình của dystrophin sẽ xuất hiện với kỹ thuật nhuộm, như thể có liên kết xung quanh các tế bào cơ cá nhân và nó đang nắm giữ chúng lại với nhau như mạng lưới. Mặc khác, một cậu bé với Duchenne sẽ thiếu sự có mặt của dystrophin và như thể thiếu sự liên kết xung quanh các tế bào cơ. Một số cá nhân có thể được tìm thấy có một số lượng trung gian của protein dystrophin. Thường thì những bé trai đó được phân loại là loạn dưỡng cơ Becker.
Xét nghiệm di truyền hay xét nghiệm adn (nhìn vào hướng dẫn di truyền của cơ thể ) trên một mẫu máu cho những thay đổi trong gen DMD có thể giúp thiết lập chẩn đoán bệnh loạn dưỡng cơ Duchenne mà không thực hiện sinh thiết cơ bắp. Thử nghiệm di truyền (xet nghiem adn, xet nghiem dna) được liên tục thay đổi, nhưng các phương pháp hiện đang được sử dụng tìm kiếm những thay đổi lớn trong các gen (mất / nhân đôi) và các phương pháp khác, mà nhìn vào các in dấu di truyền giải thích rõ ràng được tìm thấy trong các gen DMD (trình tự). Cùng hai phương pháp này có thể phát hiện các bệnh gây ra những thay đổi trong khoảng 95 % bệnh nhân. Những cá nhân không được tìm thấy có một sự thay đổi được phát hiện trong các gen DMD khi sử dụng phương pháp này và những người được chẩn đoán mắc bệnh DMD bằng sinh thiết, vẫn có một sự thay đổi trong gen của họ, nhưng đó là trong khu vực của gen mà không được kiểm tra khi sử dụng các phương pháp xét nghiệm di truyền (xet nghiem adn, xet nghiem dna). Tuy nhiên, kết quả của xét nghiệm bệnh di truyền (xét nghiệm adn, xét nghiệm dna) có thể không kết luận được một chẩn đoán của bệnh DMD và chỉ có sinh thiết cơ mới cho biết chắc chắn mức độ protein dystrophin.
Đối với các trường hợp còn lại, một sự kết hợp các kết quả lâm sàng, tiền sử gia đình, nồng độ creatine kinase máu và sinh thiết cơ bắp với các nghiên cứu dystrophin để khẳng định chẩn đoán. Creatine kinase là một enzyme thường có mặt với nồng độ cao trong các tế bào cơ bắp của cơ thể của chúng ta. Trong quá trình thoái hóa cơ bắp hoặc sự cố, các tế bào cơ bị phá vỡ mở enzyme này được giải phóng vào vào máu. Do đó nồng độ creatine kinase trong xét nghiệm máu cho biết mức độ tổn thương trong cơ bắp. Mức độ cao có thể là kết quả của nhiều nguyên nhân bao gồm chấn thương cơ cấp tính hoặc bệnh mãn tính như bệnh teo cơ Duchenne.
Cách điều trị cho bệnh loạn dưỡng cơ Duchenne là gì?
 Điều trị bệnh di truyền loạn dưỡng cơ Duchenne là nhằm vào các triệu chứng. Theo dõi tích cực bệnh giãn cơ tim với thuốc chống sung huyết được sử dụng, bao gồm cả ghép tim trong trường hợp nặng. Có thể cần các thiết bị trợ giúp cho những biến chứng hô hấp, đặc biệt là vào ban đêm. Thuốc prednisone – một steroid – được đưa ra để cải thiện sức mạnh và chức năng của các bệnh nhân DMD. Prednisone đã được chứng minh có thể kéo dài khả năng đi lại từ 2 đến 5 năm. Tuy nhiên, tác dụng phụ của prednisone bao gồm tăng cân, huyết áp cao, thay đổi hành vi và tăng trưởng chậm. Một hình thức tổng hợp của prednisilone được gọi là deflazacort được sử dụng ở châu Âu và được tin là có ít tác dụng phụ hơn so với prednisone. Một loại thuốc được gọi là cyclosporin đã được sử dụng và đã được cải thiện chức năng lâm sàng ở trẻ em, nhưng việc sử dụng chúng gây tranh cãi do bệnh cơ cyclosporin gây ra. Oxandrolone, một loại thuốc được sử dụng trong một nghiên cứu, có tác dụng tương tự như prednisone với ít tác dụng phụ. Một số phương pháp điều trị khác cũng đang được nghiên cứu, bao gồm coenzyme Q10, glutamine, pentoxifylline và PTC124 (xem nghiên cứu lâm sàng dưới đây).
Điều trị bệnh di truyền loạn dưỡng cơ Duchenne là nhằm vào các triệu chứng. Theo dõi tích cực bệnh giãn cơ tim với thuốc chống sung huyết được sử dụng, bao gồm cả ghép tim trong trường hợp nặng. Có thể cần các thiết bị trợ giúp cho những biến chứng hô hấp, đặc biệt là vào ban đêm. Thuốc prednisone – một steroid – được đưa ra để cải thiện sức mạnh và chức năng của các bệnh nhân DMD. Prednisone đã được chứng minh có thể kéo dài khả năng đi lại từ 2 đến 5 năm. Tuy nhiên, tác dụng phụ của prednisone bao gồm tăng cân, huyết áp cao, thay đổi hành vi và tăng trưởng chậm. Một hình thức tổng hợp của prednisilone được gọi là deflazacort được sử dụng ở châu Âu và được tin là có ít tác dụng phụ hơn so với prednisone. Một loại thuốc được gọi là cyclosporin đã được sử dụng và đã được cải thiện chức năng lâm sàng ở trẻ em, nhưng việc sử dụng chúng gây tranh cãi do bệnh cơ cyclosporin gây ra. Oxandrolone, một loại thuốc được sử dụng trong một nghiên cứu, có tác dụng tương tự như prednisone với ít tác dụng phụ. Một số phương pháp điều trị khác cũng đang được nghiên cứu, bao gồm coenzyme Q10, glutamine, pentoxifylline và PTC124 (xem nghiên cứu lâm sàng dưới đây).
Vật lý trị liệu được sử dụng để thúc đẩy tính di động và ngừa co rút cơ. Phẫu thuật có thể cần thiết cho sự co cứng nghiêm trọng và vẹo cột sống .
Bệnh loạn dưỡng cơ Duchenne có di truyền không?
Bệnh loạn dưỡng cơ Duchenne được di truyền lặn liên kết với NST giới tính X. Nam giới chỉ nhận một bản sao nhiễm sắc thể X từ mẹ và bản sao nhiễm sắc thể Y từ cha. Nếu nhiễm sắc thể X của họ có một đột biến gen DMD, họ sẽ mắc bệnh loạn dưỡng cơ Duchenne. Mặt khác, phụ nữ có hai bản sao của nhiễm sắc thể X. Từ khi có hai bản sao của gen này, nếu một bản sao không làm việc, người nữ có bản sao thứ hai để sản xuất các protein dystrophin. Một người phụ nữ có một sự thay đổi di truyền trong một trong hai bản sao của họ được cho là “thể mang gen” của bệnh loạn dưỡng cơ Duchenne. Thể mang gen không phát bệnh loạn dưỡng cơ Duchenne và hầu hết họ không biết rằng mình có sự đột biến trong vật chất di truyền trừ khi họ có tiền sử bệnh trong gia đình. Tuy nhiên, nghiên cứu gần đây đã chỉ ra rằng một số phụ nữ mang gen bệnh (khoảng 20 phần trăm) sẽ có triệu chứng của DMD, bao gồm yếu cơ và rối loạn tim.
Với sự di truyền lặn liên kết với X, gen đột biến (không làm việc) trên bản sao của NST của một đứa trẻ là khác nhau ở nam và nữ.
Những người phụ nữ mang các bản sao đột biến gen có 50% sẽ truyền cho con khi mai thai. Do đó, có 25% tỉ lệ việc có một đứa trẻ bị ảnh hưởng với DMD (ví dụ , 50% bé trai có xác suất mắc bệnh DMD và 50% của các bé gái sẽ là những người mang gen bệnh). Xác suất một người phụ nữ đã có một con trai bị ảnh hưởng (và không có tiền sử gia đình) của thể mang gen DMD đột biến là khoảng 2/3. Tuy nhiên, ở một phần ba còn lại của các cá nhân DMD, sự thay đổi trong gen dystrophin là một sự thay đổi di truyền mới và khoảng 10% của đột biến mới này là do khảm tuyến sinh dục. Khảm sinh dục nghĩa là một cá nhân có hai hay nhiều quần thể tế bào khác nhau trong cấu trúc di truyền trong trứng hoặc tinh trùng của họ .
Nam giới thừa kế hoặc được sinh ra với một bản sao đột biến của gen DMD sẽ mắc bệnh DMD vì họ có một nhiễm sắc thể Y và không có nhiễm sắc thể X thứ hai. Nếu một người đàn ông với DMD có con, tất cả các con gái của ông sẽ là thể mang gen và không ai trong số con trai của ông sẽ bị ảnh hưởng.
Hiện nay lựa chọn sinh sản khác nhau có sẵn cho các gia đình. Các tùy chọn định kiến bao gồm MicroSort là một công nghệ có thể tách tinh trùng chứa nhiễm sắc thể X cho phép tăng khả năng có một nữ. Lựa chọn sinh sản thứ hai là chẩn đoán di truyền preimplantation (PGD) (hay xét nghiệm adn), đó là một kỹ thuật có thể cho phép các tế bào của trứng đã thụ tinh được thử nghiệm để xác định xem nó có hay không chứa đột trong gen DMD và sau đó cấy những quả trứng nào không mang gen bệnh. Các tùy chọn xét nghiệm bao gồm: Lấy mẫu lông nhung màng đệm (CVS) và chọc ối, phân tích các tế bào lấy mẫu có nguồn gốc từ phôi đang phát triển.
Một số tùy chọn xét nghiệm tiền sản cho thai khi nguy cơ có DMD đột biến gây bệnh đã được xác định trong một thành viên gia đình, hoặc nếu thông tin đánh dấu di truyền liên kết, đã được xác định bởi xet nghiem adn (xet nghiem dna)
Nguồn: National Human Genome Research Institute